9. Variant calling¶
9.1. Preface¶
In this section we will use our genome assembly based on the ancestor and call genetic variants in the evolved line [NIELSEN2011]. “Call variants” is the terminology we use when we are looking for mutations that have occurred when we compare the sequence from one clone (or individual) to another. A “variant” might be a single nucleotide substitution (e.g. T –> C), or it could be a deletion, a repeat expansions, etc. The aim in “variant calling” is to figure out how your sequence differs from the reference sequence that you have.
For humans, this is usually done to infer SNPs that exist between individuals. Here, we are doing it to find mutations that have occurred during experimental evolution.
9.2. Overview¶
The part of the workflow we will work on in this section can be viewed in Fig. 9.1.
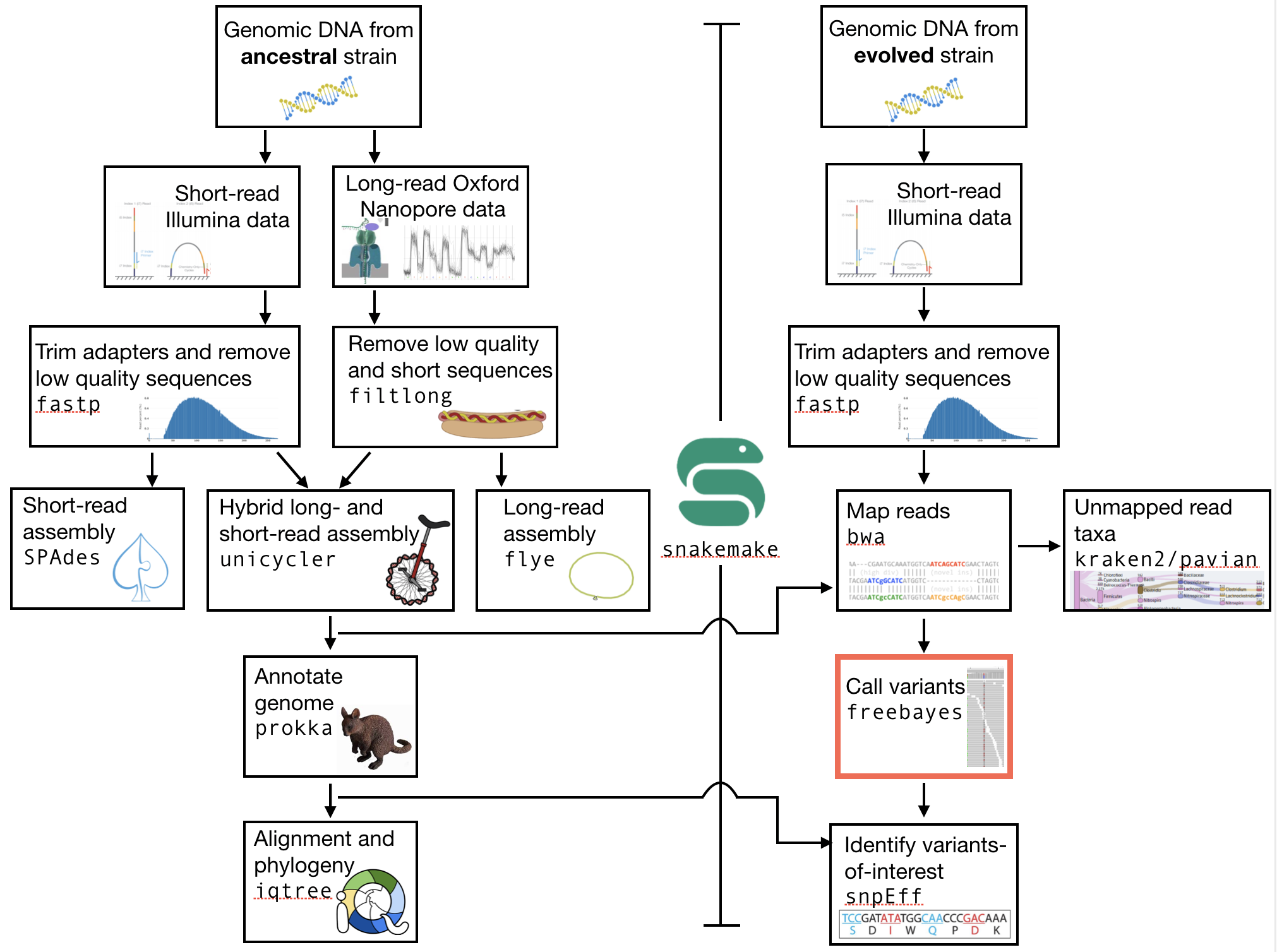
Fig. 9.1 The part of the workflow we will work on in this section marked in red.¶
9.3. Learning outcomes¶
After studying this tutorial section you should be able to:
Use tools to call variants based on a reference genome.
Be able to describe what influences the calling of variants.
9.4. Installing the necessary software¶
Tools we are going to use in this section and how to intall them if you not have done it yet.
# activate the env
conda activate ngs
# Install these tools into the conda environment
# if not already installed
conda install samtools
conda install bamtools
conda install freebayes
conda install bedtools
conda install vcflib
conda install rtg-tools
conda install bcftools
9.5. Preprocessing¶
Attention
All the variant-calling that we will do must be done on the unicycler reference that you have assembled.
We first need to make an index of our reference genome as this is required by the SNP caller.
Given an assembly file in fasta-format, e.g. assembly.fasta which is located in the directory, use SAMtools to do this:
Attention
Remember to replace the file names and directory names below with the names that are relevant for your files (e.g. results/assembly.fasta may need to be replaced with the path to your specific assembly).
samtools faidx results/assembly.fasta
This command will output a new file with the extension .fai. However, this output is not used within the command itself. Thus, while you should specify this file as output for this rule, you should not use it when typing the actual commands in the command section. In addition, you will need to specify this .fai output file as input for bcftools and freebayes rules, but again, you will not actually use the input in the command section.
Furthermore we need to pre-process our mapping files a bit further and create a bam-index file (.bai) for the bam-file we want to work with:
bamtools index -in results/my_mapped_sorted_dedup_concordant.q20.bam
As above for the faidx command, there is an output for this command, which is a .bai file. However, this file is never specified in the actual command, but needs to be specified in the output for your rule. And again, as above, this file needs to specificed as input for your bcftools or freebayes rules.
If you would like you can also create a new directory for the variants (e.g. variants).
9.6. Calling variants¶
9.6.1. bcftools mpileup¶
We use the sorted filtered bam-file that we produced in the mapping step before. Note that below we specify the .bcf output as being produced using bcftools by explicitly adding bcftools to the file name.
# We first pile up all the reads and then call
# variants using the pipe | operator
bcftools mpileup -f results/assembly.fasta my_sorted_dedup_q20.bam | bcftools call -v -m -Ob -o my_variant_calls_bcftools.bcf
This is a rather complicated instruction, which is partly due to
the fact that there has been a very
recent change from the tool used previously for this step, samtools mpileup.
With bcftools mpileup we use the pipe (|) operator
because we have no need ever for the intermediate output,
and instead feed the output of bcftools mpileup directly to bcftools call. There are several options that we invoke, explained below:
BCFtools mpileup parameter:
-f FILE: faidx indexed reference sequence file
BCFtools call parameters:
-v: output variant sites only-m: alternative model for multiallelic and rare-variant calling-o: output file-name-Ob: output type: binary compressed VCF
9.6.2. Freebayes¶
As an alternative we can do some variant calling with another tool called freebayes. In fact one reason to do so would be to compare the results of bcftools and freebayes, and (for example) focus only on variant calls that are made by both tools.
Given a reference genome assembly file in fasta-format, e.g. assembly.fasta and the index in .fai format and a mapping file (.bam file) and a mapping index (.bai file), we can call variants with freebayes like so (it is probably agood idea to note the output by specifying freebayes in the file name:
# Now we call variants and pipe the results into a new file
freebayes -f results/assembly.fasta my_sorted_dedup_q20.bam > my_sorted_dedup_q20_freebayes.vcf
9.7. Post-processing¶
9.7.1. Understanding the output files (.vcf)¶
Lets look at a vcf-file:
# first 10 lines, which are part of the header
# you know how to do this but I write
# it out anyway
head variants/evolved-6.mpileup.vcf
Lets look at the variants using less:
# you will need to scroll a little
# after using less to get to the variant calls
less myvariants.bcftools.vcf
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT H8_sorted.bam
1 59501 . C A 228 . DP=130;VDB=0.0235953;SGB=-0.693147;RPB=0.130017;MQB=3.91681e-08;MQSB=9.58804e-08;BQB=0.0391486;MQ0F=0.415385;AC=2;AN=2;DP4=39,0,29,27;MQ=16 GT:PL 1/1:255,17,0
1 59593 . A C 228 . DP=120;VDB=0.845548;SGB=-0.693147;RPB=0.612735;MQB=1.17223e-07;MQSB=8.69064e-05;BQB=0.00321345;MQ0F=0.525;AC=2;AN=2;DP4=39,7,27,24;MQ=12 GT:PL 1/1:255,11,0
1 59614 . A G 228 . DP=119;VDB=0.734093;SGB=-0.693147;RPB=0.902247;MQB=3.43515e-06;MQSB=0.0567731;BQB=0.0325125;MQ0F=0.537815;AC=2;AN=2;DP4=35,10,30,18;MQ=10 GT:PL 1/1:255,11,0
If you look carefully, you might notice that your variant calls are not spread evenly throughout the genome. This is because there are certain error-prone locations in your assembly. These are areas in which the assembly is not correct (or, is not likely to be correct), and in these places, many variants get called. The fields in a vcf-file are described in the table (Table 9.1) below:
Col |
Field |
Description |
|---|---|---|
1 |
CHROM |
Chromosome name |
2 |
POS |
1-based position. For an indel, this is the position preceding the indel. |
3 |
ID |
Variant identifier. Usually the dbSNP rsID. |
4 |
REF |
Reference sequence at POS involved in the variant. For a SNP, it is a single base. |
5 |
ALT |
Comma delimited list of alternative seuqence(s). |
6 |
QUAL |
Phred-scaled probability of all samples being homozygous reference. |
7 |
FILTER |
Semicolon delimited list of filters that the variant fails to pass. |
8 |
INFO |
Semicolon delimited list of variant information. |
9 |
FORMAT |
Colon delimited list of the format of individual genotypes in the following fields. |
10+ |
Sample(s) |
Individual genotype information defined by FORMAT. |
9.7.2. Statistics¶
Now we can use it to do some statistics and filter our variant calls.
For example, we can get some quick stats with rtg vcfstats:
rtg vcfstats my_variant_calls_freebayes.vcf
Example output from rtg vcfstats:
Location : variants/evolved-6.mpileup.vcf.gz
Failed Filters : 0
Passed Filters : 516
SNPs : 399
MNPs : 0
Insertions : 104
Deletions : 13
Indels : 0
Same as reference : 0
SNP Transitions/Transversions: 1.87 (286/153)
Total Het/Hom ratio : 3.20 (393/123)
SNP Het/Hom ratio : 8.98 (359/40)
MNP Het/Hom ratio : - (0/0)
Insertion Het/Hom ratio : 0.30 (24/80)
Deletion Het/Hom ratio : 3.33 (10/3)
Indel Het/Hom ratio : - (0/0)
Insertion/Deletion ratio : 8.00 (104/13)
Indel/SNP+MNP ratio : 0.29 (117/399)
However, we can also run BCFtools to extract more detailed statistics about our variant calls:
bcftools stats -F results/assembly.fasta -s - my_variant_calls_freebayes.vcf > my_variant_calls_freebayes.vcf.stats
-s -: list of samples for sample stats, “-” to include all samples-F FILE: faidx indexed reference sequence file to determine INDEL context
Now we can take the stats and make some plots (e.g. Fig. 9.2) which are particular of interest if having multiple samples, as one can easily compare them. However, most of you are only working with one here:
plot-vcfstats -p freebayes my_variant_calls_freebayes.vcf.stats
-p: The output files prefix, add a slash at the end to create a new directory.
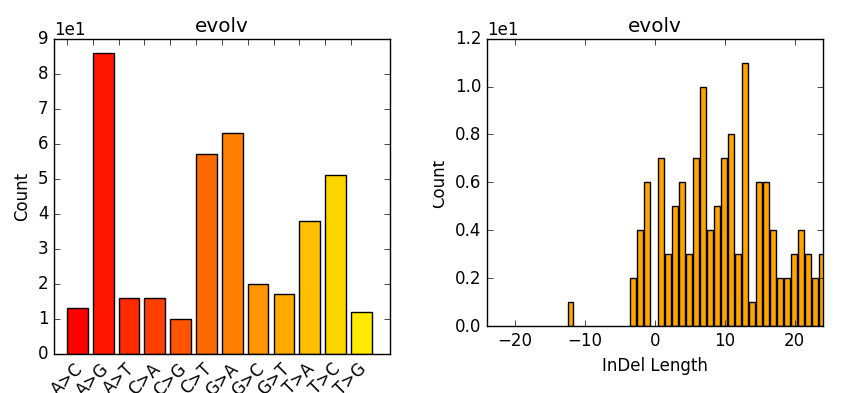
Fig. 9.2 Example of plot-vcfstats output.¶
Attention
You may find that you get an error saying you do not have tectonic or pdflatex in your $PATH. If so, you will need to install tectonic: conda install -c conda-forge tectonic.
9.7.3. Variant filtration¶
Variant filtration is a big topic in itself [OLSEN2015].
There is no consens yet and research on how to best filter variants is ongoing. In addition (and rather surprisingly), the two methods that we have used to call variants, vcftools mpileup and freebayes differ considerably the quality scores that they assign. vcftools assigns a maximum of 228; freebayes has no maximum, and you will see that many scores are above 1000.
We will do some simple filtration procedures here. For one, we can filter out low quality reads.
Here, we only include variants that have quality > 220.
# use rtg vcffilter
rtg vcffilter -Z -q 220 -i my_variant_calls_bcftools.vcf -o my_variant_calls_bcftools.q220.vcf
-i FILE: input file-o FILE: output file-Z: do not compress the output-q FLOAT: minimal allowed quality in output.
Quick stats for the filtered variants:
# look at stats for filtered
rtg vcfstats my_variant_calls_bcftools.q220.vcf
freebayes adds some extra information to the vcf-files it creates. This allows for some more detailed filtering. This strategy will not work on the |vcftools| mpileup called variants Here we filter, based on some recommendations from the developer of freebayes:
vcffilter -f "QUAL > 1 & QUAL / AO > 10 & SAF > 0 & SAR > 0 & RPR > 1 & RPL > 1" my_variant_calls_freebayes.vcf > my_variant_calls_freebayes.quality.vcf
QUAL > 1: removes really bad sitesQUAL / AO > 10: additional contribution of each obs should be 10 log units (~ Q10 per read)SAF > 0 & SAR > 0: reads on both strandsRPR > 1 & RPL > 1: at least two reads “balanced” to each side of the site
9.7.4. Transition Transversion ToDo¶
Todo
Look at the statistics. One ratio that is mentioned in the statistics is transition transversion ratio (ts/tv). Explain what this ratio is and why the observed ratio makes sense.
This strategy used here will do for our purposes. However, several more elaborate filtering strategies have been explored, e.g. here.
References
- NIELSEN2011
Nielsen R, Paul JS, Albrechtsen A, Song YS. Genotype and SNP calling from next-generation sequencing data. Nat Rev Genetics, 2011, 12:433-451
- OLSEN2015
Olsen ND et al. Best practices for evaluating single nucleotide variant calling methods for microbial genomics. Front. Genet., 2015, 6:235.