8. Taxonomic investigation¶
8.1. Preface¶
We want to investigate if there are sequences of other species in our collection of sequenced DNA pieces. We hope that most of them are from our species that we try to study, i.e. the DNA that we have extracted and amplified. This might be a way of quality control, e.g. have the samples been contaminated (Fig. 8.1)? Lets investigate if we find sequences from other species in our sequence set.

Fig. 8.1 Gotta be careful.¶
We will use the tool Kraken2 to assign taxonomic classifications to our sequence reads. Let us see if we can id some sequences from other species.
Note
You will encounter some To-do sections at times. Write the solutions and answers into a text-file.
8.2. Overview¶
The part of the workflow we will work on in this section can be viewed in Fig. 8.2.
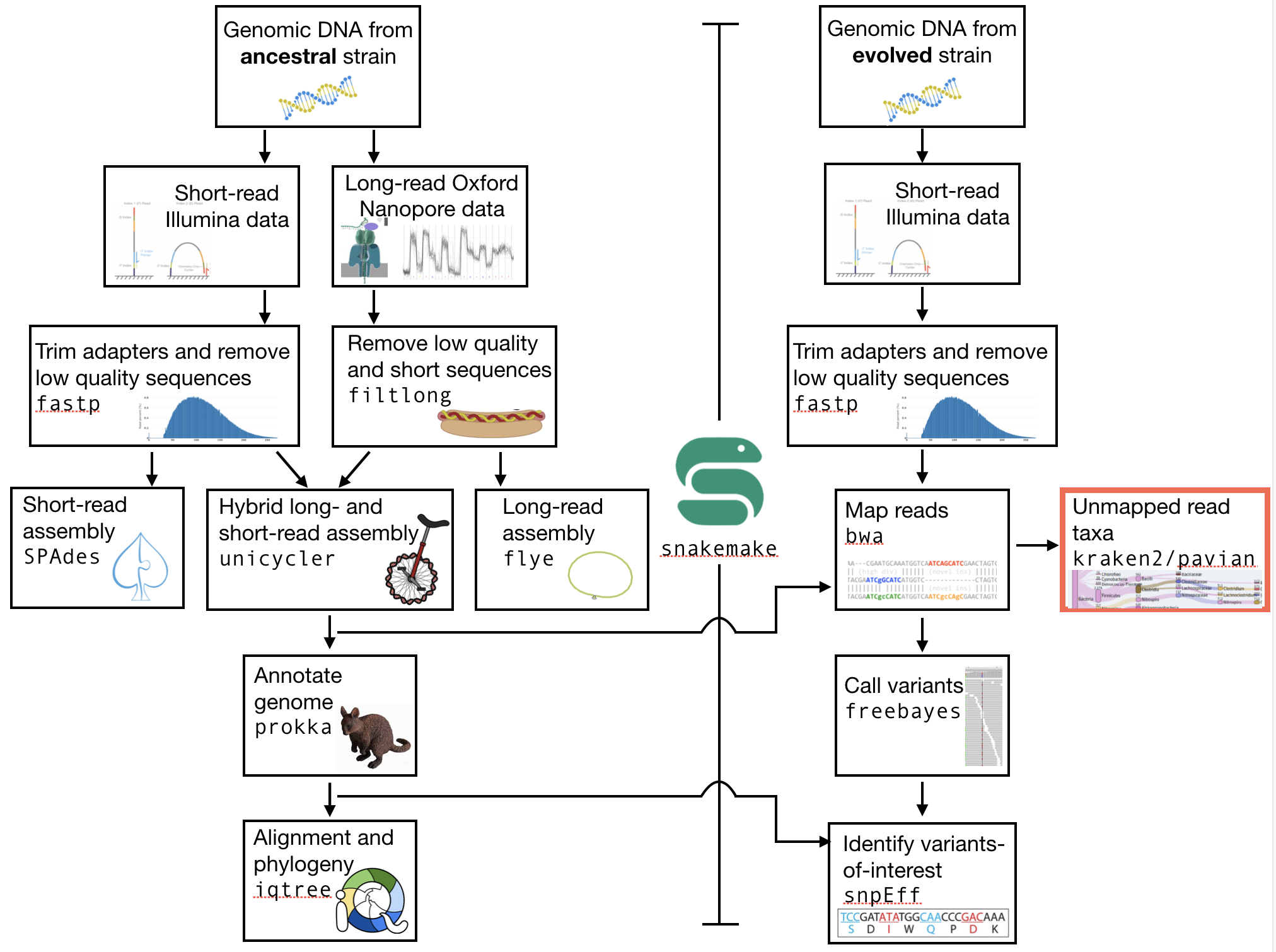
Fig. 8.2 The part of the workflow we will work on in this section marked in red.¶
8.3. Before we start¶
You should have a Snakefile for the other stepsof this workflow, including the previous step, read mapping. You will also have (minimally) two directories: your data diirectory (with subdirectories), and your results directory (perhaps woth subdirectories).
Today we will first do the steps without incorporating them into the Snakefile. After you have completed the steps, you can incorporate them into the Snakefile.
8.4. Kraken2¶
We will be using a tool called Kraken2 [WOOD2014]. This tool uses k-mers to assign a taxonomic labels in form of NCBI Taxonomy to the sequence (if possible). The taxonomic label is assigned based on similar k-mer content of the sequence in question to the k-mer content of reference genome sequence. The result is a classification of the sequence in question to the most likely taxonomic label. If the k-mer content is not similar to any genomic sequence in the database used, it will not assign any taxonomic label.
8.4.1. Installation¶
Use conda in the same fashion as before to install Kraken2. However, we are going to install kraken into its own environment:
conda create --yes -n kraken kraken2 bracken
conda activate kraken
Now we create a directory where we are going to do the analysis and we will change into that directory too.
# make sure you are in your analysis root folder
cd ~/genome_analysis
# create a kraken directory
# of course I leave this for you to do
Now we need to create or download or find a Kraken2 database that can be used to assign the taxonomic labels to sequences.
Fortunately, there is one conveniently located on the server. It can be found here: /home2/gs/data/minikraken_v2:
8.4.1.1. Database ToDo¶
Todo
The “minikraken2” database was created from bacteria, viral and archaea sequences. What are the implications for us when we are trying to classify our sequences?
8.4.2. Usage¶
Now that we have installed Kraken2 and downloaded and extracted the minikraken2 database, we can attempt to investigate the sequences we got back from the sequencing provider for other species as the one it should contain.
We call the Kraken2 tool and specify the database and fasta-file with the sequences it should use. To investigate the general command structure simply type kraken2:
# see how kraken2 works and what the
# arguments are
kraken2
Need to specify input filenames!
Usage: kraken2 [options] <filename(s)>
Options:
--db NAME Name for Kraken 2 DB
(default: none)
--threads NUM Number of threads (default: 1)
--quick Quick operation (use first hit or hits)
--unclassified-out FILENAME
Print unclassified sequences to filename
Note that the input you are using is fastq.
In addition, we are dealing with paired-end data, which we can tell Kraken2 using the switch --paired. Choose the unmapped read file that you have created as input for kraken2. Also note that you should output a report file (use --report FILENAME). Also note that the output of kraken2 goes to the command line (standard out), so you should use the redirect to output to a text file. You can name this file something like myfile.kraken as it is not quite a normal text file, and this indicates that it is formatted for kraken2.
The resulting content of your kraken2 file should look similar to the following example:
C 7001326F:121:CBVVLANXX:1:1105:2240:12640 816 251 816:9 171549:5 816:5 171549:3 2:2 816:5 171549:4 816:34 171549:8 816:4 171549:2 816:10 A:35 816:10 171549:2 816:4 171549:8 816:34 171549:4 816:5 2:2 171549:3 816:5 171549:5 816:9
C 7001326F:121:CBVVLANXX:1:1105:3487:12536 1339337 202 1339337:67 A:35 1339337:66
U 7001326F:121:CBVVLANXX:1:1105:5188:12504 0 251 0:91 A:35 0:91
U 7001326F:121:CBVVLANXX:1:1105:11030:12689 0 251 0:91 A:35 0:91
U 7001326F:121:CBVVLANXX:1:1105:7157:12806 0 206 0:69 A:35 0:68
Each sequence classified by Kraken2 results in a single line of output. Output lines contain five tab-delimited fields; from left to right, they are:
C/U: one letter code indicating that the sequence was either classified or unclassified.The sequence ID, obtained from the FASTA/FASTQ header.
The taxonomy ID Kraken2 used to label the sequence; this is 0 if the sequence is unclassified and otherwise should be the NCBI Taxonomy identifier.
The length of the sequence in bp.
A space-delimited list indicating the lowest common ancestor (in the taxonomic tree) mapping of each k-mer in the sequence. For example,
562:13 561:4 A:31 0:1 562:3would indicate that:the first 13 k-mers mapped to taxonomy ID #562
the next 4 k-mers mapped to taxonomy ID #561
the next 31 k-mers contained an ambiguous nucleotide
the next k-mer was not in the database
the last 3 k-mers mapped to taxonomy ID #562
8.4.3. Investigate the taxa¶
We can use the webpage NCBI TaxIdentifier to quickly get the names to the taxonomy identifier. However, this is impractical as we are dealing potentially with many sequences. Kraken2 has some scripts that help us understand our results better.
Because you (hopefully) used the Kraken2 switch --report FILE, you also have a sample-wide report of all taxa found.
This is much better to get an overview what was found.
The first few lines of an example report are shown below.
83.56 514312 514312 U 0 unclassified
16.44 101180 0 R 1 root
16.44 101180 0 R1 131567 cellular organisms
16.44 101180 2775 D 2 Bacteria
13.99 86114 1 D1 1783270 FCB group
13.99 86112 0 D2 68336 Bacteroidetes/Chlorobi group
13.99 86103 8 P 976 Bacteroidetes
13.94 85798 2 C 200643 Bacteroidia
13.94 85789 19 O 171549 Bacteroidales
13.87 85392 0 F 815 Bacteroidaceae
The output of kraken-report is tab-delimited, with one line per taxon. The fields of the output, from left-to-right, are as follows:
Percentage of reads covered by the clade rooted at this taxon
Number of reads covered by the clade rooted at this taxon
Number of reads assigned directly to this taxon
A rank code, indicating (U)nclassified, (D)omain, (K)ingdom, (P)hylum, (C)lass, (O)rder, (F)amily, (G)enus, or (S)pecies. All other ranks are simply “-“.
The indented scientific name
8.4.4. Bracken¶
Bracken stands for Bayesian Re-estimation of Abundance with KrakEN, and is a statistical method that computes the abundance of species in DNA sequences from a metagenomics sample [LU2017]. Crudely, this relies on the inferred frequencies of different taxa to gain confidence (or lose confidence) that other taxa are present or absent. For example, if there are 1,000 reads that match E. coli and only one that matches the closely related taxon Shigella, then the Shigella read might be assigned as belonging to E. coli.
Combined with the Kraken classifier, Bracken will produces more accurate species- and genus-level abundance estimates than Kraken2 alone.
The use of Bracken subsequent to Kraken2 is optional - but it might improve on the Kraken2 results.
We installed Bracken already together with Kraken2 above, so it should be ready to be used. We also downloaded the Bracken files together with the minikraken2 database above, so we are good to go.
Now, we can use Bracken on the Kraken2 results to improve them.
The general structure of the Bracken command look like this:
# of course you can name the output file as you please
bracken -d PATH_TO_DB_DIR -i kraken2.report -o bracken.species.txt -l S
-l S: denotes the level we want to look at.Sstands for species - but other levels are available.-d PATH_TO_DB_DIR: specifies the path to the Kraken2 database that should be used.
Go ahead and apply Bracken to your kraken2 output file (not the report file):
The species-focused result-table looks similar to this:
name taxonomy_id taxonomy_lvl kraken_assigned_reads added_reads new_est_reads fraction_total_reads
Streptococcus sp. oral taxon 431 712633 S 2 0 2 0.00001
Neorhizobium sp. NCHU2750 1825976 S 0 0 0 0.00000
Pseudomonas sp. MT-1 150396 S 0 0 0 0.00000
Ahniella affigens 2021234 S 1 0 1 0.00000
Sinorhizobium sp. CCBAU 05631 794846 S 0 0 0 0.00000
Cohnella sp. 18JY8-7 2480923 S 1 0 1 0.00000
Bacillus velezensis 492670 S 4 4 8 0.00002
Actinoplanes missouriensis 1866 S 2 8 10 0.00002
The important column is the new_est_reads, which gives the newly estimated reads.
8.5. Visualisation (Pavian)¶
We use the tool Pavian to create an interactive visualisation of the taxa content of our sample [ONDOV2011].
Fig. 8.3 shows an example (albeit an artificial one) snapshot of the Pavian visualisation.
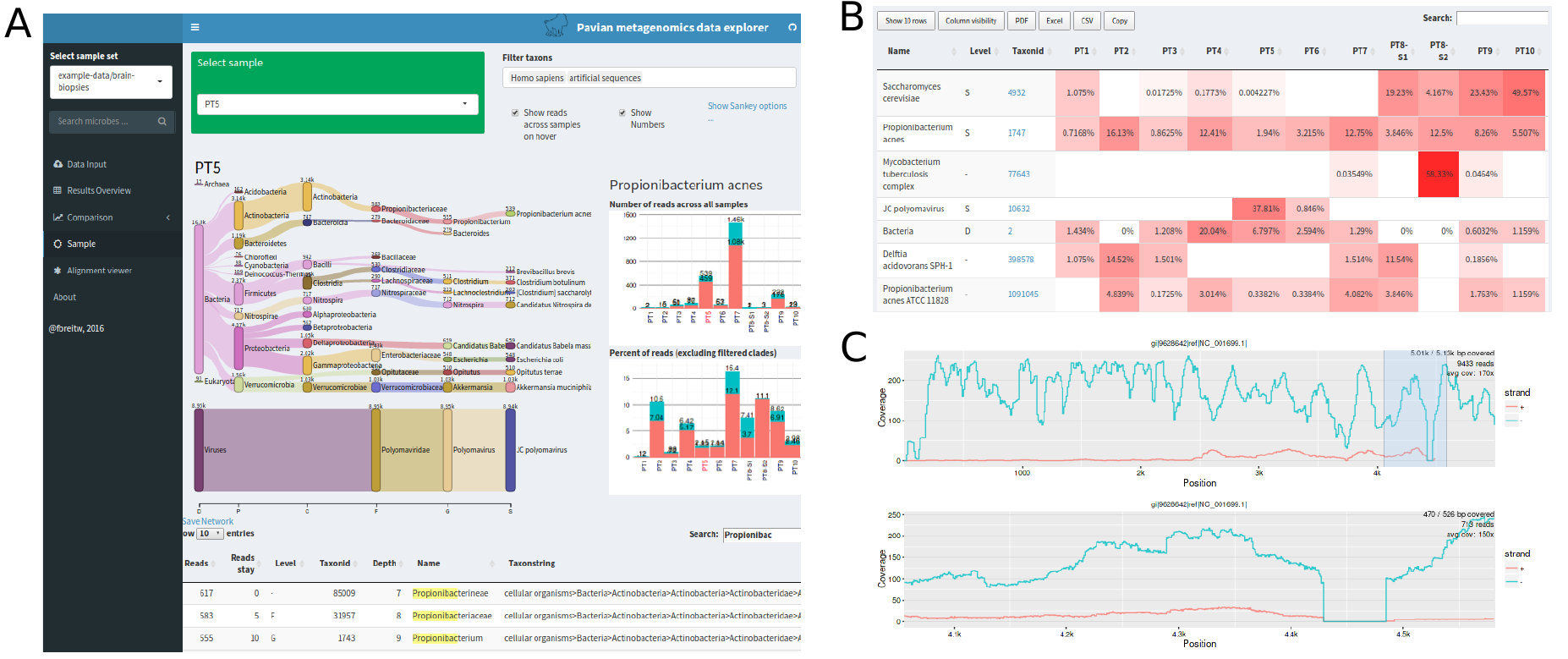
Fig. 8.3 Example of a Pavian interactive report.¶
Fortunately for us, Pavian can run as an interactive web page, and can directly import kraken2 and bracken reports. This means that you will only have to rsync or scp the kraken (or bracken) report than you have created onto your desktop, and from there you can access the Pavian website. You can reach this by using the link, or by typing this link into your browser: https://fbreitwieser.shinyapps.io/pavian/.
Once on the Pavian website, you should see the option to upload your kraken2 report. Go ahead and do so. This willl result in an interactive report.
Attention
If you incorporate the kraken2 analysis into your Snakefile then this will result in several kraken2 reports being created. It is possible to download all of these and upload them simultaneously to the Pavian web server. In this way, you can see how the different samples might differ in their contaminants (or not).
References
- KIM2017
Kim D, Song L, Breitwieser FP, Salzberg SL. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 2016 Dec;26(12):1721-1729
- LU2017
Lu J, Breitwieser FP, Thielen P, Salzberg SL. Bracken: estimating species abundance in metagenomics data. PeerJ Computer Science, 2017, 3:e104, doi:10.7717/peerj-cs.104
- ONDOV2011
Ondov BD, Bergman NH, and Phillippy AM. Interactive metagenomic visualization in a Web browser. BMC Bioinformatics, 2011, 12(1):385.
- WOOD2014
Wood DE and Steven L Salzberg SL. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biology, 2014, 15:R46. DOI: 10.1186/gb-2014-15-3-r46.